乳腺癌作为威胁全球女性健康的头号恶性肿瘤,约70%病例呈现雌激素受体α(ERα)阳性特征。尽管临床上基于ERα信号通路的内分泌疗法已显著改善患者生存,但高达半数患者因获得性耐药面临治疗失败的风险。武汉大学周海兵教授课题组近年来聚焦于乳腺癌内分泌治疗耐药这一关键难题,在靶向ERα降解剂药物研究领域取得了系列进展,代表性成果包括:共价选择性雌激素受体降解剂(cSERD)(Acta Pharm. Sin. B. 2023, 13, 4963-4982)、ERα靶向蛋白水解靶向嵌合体(ERα-PROTAC)降解剂(J. Med. Chem. 2023, 66, 6631-6651),以及ERα/ARO(芳构化酶)双靶点PROTAC降解剂(J. Med. Chem. 2024, 67, 8913-8931)等。然而乳腺癌耐药机制错综复杂,其中ERα配体结合域(LBD)突变(如Y537S、D538G)与共激活因子如SRC-3过表达构成核心挑战。近日,武汉大学周海兵教授团队在药物化学顶级期刊Journal of Medicinal Chemistry连续发表两项研究进展,针对ERα信号通路转导特点,分别通过靶向下游信号枢纽SRC-3和ERα共激活因子结合位点,设计了两类新型PROTAC降解剂,为克服耐药开辟新路径。
进展一:拦截ERα下游通路—靶向共激活因子SRC-3降解剂
SRC-3 作为致癌共激活因子,在多种癌症中扮演着重要角色,尤其是作为ERα信号通路下游的关键放大器,对肿瘤的发生、发展和转移起到了关键的促进作用,这也为癌症的治疗提供了潜在的靶点和研究方向。研究发现,SRC-3不仅在与ERα的结合中促进耐药,还可通过激活p53、PELP1等非ER通路驱动肿瘤进展。基于此,团队以高选择性SRC-3抑制剂SI-2为弹头,通过PROTAC设计策略,构建出首个靶向SRC-3的PROTAC降解剂BY13。该化合物在1 μM浓度下可高效降解SRC-3(71%),并同步下调ERα水平达85%,这种“一石二鸟”效应可能源于SRC-3缺失破坏了对ERα的翻译后修饰保护机制。在耐药模型中,BY13展现出强效的活性,对Y537S突变细胞抑制活性(IC50 = 0.003 μM)显著优于临床治疗药物氟维司群,并且在他莫昔芬耐药的LCC-2细胞中仍保持0.35 μM的高效抑制。机制研究表明,BY13通过诱导SRC-3-BY13-CRBN三元复合物形成,触发泛素-蛋白酶体依赖的降解,并特异性阻滞细胞周期于G1期。尤其在LCC-2耐药小鼠模型中, BY13表现出优异的肿瘤抑制活性。相关成果已发表在Journal of Medicinal Chemistry(2025, 68, 11516-11542; https://doi.org/10.1021/acs.jmedchem.5c00425)。该研究工作由博士生梁锦森、王丹丹与吴亦何等共同完成,药学院周海兵教授、医学研究院李姝教授和舒红兵院士为共同通讯作者。
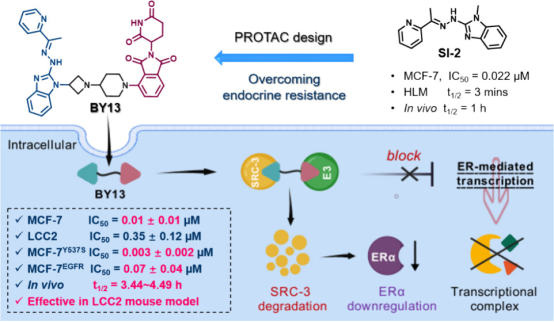
进展二:规避传统内分泌治疗药物结合位点—靶向ERα共激活因子结合位点PROTAC
与靶向ERα下游信号通路的策略不同,此项研究聚焦于耐药机制的核心本源——ERα突变导致的自身功能异常。团队另辟蹊径,绕过易突变的经典ERα配体结合域(LBD),设计靶向共激活因子结合位点(CBS)的PROTAC。通过系统优化最终获得先导化合物CP03,该分子可直接阻断ERα转录信号,同时劫持泛素-蛋白酶体系统(UPS)诱导 ERα降解。该方案不仅有望解决 ERα 突变体(ERαMUT)介导的非配体依赖激活引发的内分泌耐药问题,同时可以克服目前共激活因子结合抑制剂(CBIs)亲和力弱、活性低及无降解活性等不足。相较于ERα野生型细胞,CP03在ERαMUT及耐药乳腺癌细胞系中展现出更强的抗增殖活性和靶蛋白降解能力。通过SPR与分子动力学模拟证实,CP03可与ERα形成稳定的ERα-CP03-VHL三元复合物,驱动ERα降解。进一步机制研究表明CP03对ERα的降解展现出高效选择性,并且不受内源性配体雌激素的干扰。在动物实验中,CP03在2 mg/kg剂量下对耐药肿瘤的抑制效果优于氟维司群,为“非LBD依赖”的ERα靶向治疗提供首个例证。相关成果已在线发表在Journal of Medicinal Chemistry(https://doi.org/10.1021/acs.jmedchem.5c00495)。该研究由博士生王禹博、谢宝花等共同完成,通讯作者为周海兵教授与董春娥教授。
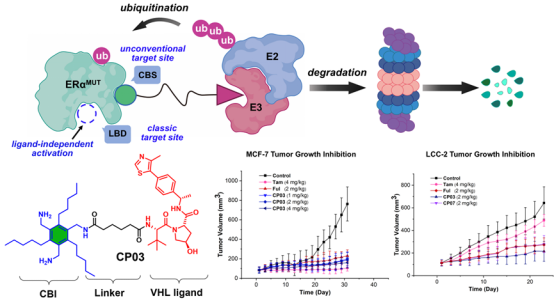
这两项概念验证性(Proof-of-Concept)研究工作虽靶点各异,却在机制层面呈现互补性。SRC-3降解剂BY13从信号通路下游“釜底抽薪”,阻断ERα转录复合物的最终组装;而ERα-CBS靶向降解剂CP03则从上游直接破坏ERα与共激活因子的结合基础。同时,两者均巧妙规避了传统靶向配体结合域(LBD)药物的不足,即BY13和CP03不受ERα突变影响;此外,二者共同验证了PROTAC技术在克服“不可成药”靶点上的独特优势:即使传统抑制剂难以结合的蛋白相互作用界面(如CBS)或无催化功能的支架蛋白(如SRC-3),亦可被PROTAC靶向并有效降解。
以上研究工作得到了国家自然科学基金、国家重点研发计划项目和教育部免疫与代谢前沿科学中心的支持。

